In Prasco v. Medicis Pharmaceutical (07-1524), Prasco tried to get a declaratory judgment that one of its products did not infringe various patents owned by Medicis Pharmaceutical Corp. and Imaginative Research Associates. The district court dismissed the action for lack of jurisdiction, concluding that Prasco’s complaint failed to establish a case or controversy under Article III of the Constitution.
Medicis markets a benzoyl peroxide cleansing product TRIAZ®, which is marked as being covered by U.S. Patent Nos. 5,648,389; 5,254,334; 5,409,706; and 5,632,996. Prasco, which makes a generic benzoyl peroxide cleansing product OSCIONâ„¢, wanted a declaratory judgment that OSCIONâ„¢ did not infringe the patents
This problem can also be viewed through one of the prongs of the ripeness doctrine, whether the complained-of-conduct has an “immediate and substantial impact” on the plaintiff such that withholding court consideration would cause hardship to the plaintiff. Article III courts cannot issue advisory opinions.
Prasco sent a sample of OSCIONâ„¢ and an ingredient list to Medicis and Imaginative Research Associates and requested a covenant not to sue under the four patents. Medicis and Imaginative Research Associates returned the favor by pressing for a motion to dismiss.
The Supreme Court, in MedImmune v. Genentech, reaffirmed that the proper test for subject matter jurisdiction in declaratory judgment actions is “whether the facts alleged, under all the circumstances, show that there is a substantial controversy, between the parties having adverse legal interests, of sufficient immediacy and reality to warrant the issuance of a declaratory judgment.”
The district court applied the Federal Circuit’s reasonable apprehension of suit test and found that Prasco had not alleged a case or controversy noting that even if MedImmune had overruled the reasonable apprehension of suit test, it would still conclude that there was no case or controversy because there was “no definite and concrete dispute that touches the legal relations of these parties.”
The Declaratory Judgment Act provides:
In a case of actual controversy within its jurisdiction . . . any court of the United States, upon the filing of an appropriate pleading, may declare the rights and other legal relations of any interested party seeking such declaration, whether or not further relief is or could be sought.
The Declaratory Judgment Act provides a remedy available only if the court has jurisdiction from some other source, limited by Article III of the Constitution, which restricts federal judicial power to the adjudication of “Cases” or “Controversies.”
Per MedImmune, for there to be a case or controversy under Article III, the dispute must be “definite and concrete, touching the legal relations of parties having adverse legal interests,” “real and substantial,” and “admi[t] of specific relief through a decree of a conclusive character, as distinguished from an opinion advising what the law would be upon a hypothetical state of facts.”
The standard is whether “the facts alleged, under all the circumstances, show that there is a substantial controversy, between parties having adverse legal interests, of sufficient immediacy and reality to warrant the issuance of a declaratory judgment.”
Prior to MedImmune, the federal circuit had generally required that a declaratory judgment plaintiff in a patent dispute demonstrate (1) conduct by the patentee that created a “reasonable apprehension” of suit on the part of the declaratory judgment plaintiff and (2) present activity by the declaratory judgment plaintiff that could constitute infringement or “meaningful preparation” to conduct potentially infringing activity.
However, in MedImmune, the Supreme Court found that requiring a reasonable apprehension of suit conflicted with the Court’s precedent. While the Supreme Court rejected the reasonable apprehension of suit test as the sole test for jurisdiction, it did not completely do away with the relevance of a reasonable apprehension of suit. Rather, following MedImmune, proving a reasonable apprehension of suit is one of multiple ways that a declaratory judgment plaintiff can satisfy the more general all-the-circumstances test to establish that an action presents a justiciable Article III controversy.
Here, the Federal Circuit did not think that the facts created the necessary elements of a justiciable Article III controversy:
Considering the totality of the circumstances, Prasco has not alleged a controversy of sufficient “immediacy and reality” to create a justiciable controversy. This “immediacy and reality” inquiry can be viewed through the lens of standing. To satisfy standing, the plaintiff must allege (1) an injury-in-fact, i.e., a harm that is “‘concrete’ and actual or imminent, not ‘conjectural’ or ‘hypothetical,’” (2) that is “fairly traceable” to the defendant’s conduct, and (3) redressable by a favorable decision.
As Prasco acknowledges, MedImmune does not change our long-standing rule that the existence of a patent is not sufficient to establish declaratory judgment jurisdiction. See Capo, Inc. v. Dioptics Med. Prods. Inc., 387 F.3d 1352, 1355 (Fed. Cir. 2004) (“More is needed than knowledge of . . . an adversely held patent.”). The mere existence of a potentially adverse patent does not cause an injury nor create an imminent risk of an injury; absent action by the patentee, “a potential competitor . . . is legally free to market its product in the face of an adversely-held patent.”
Prasco argued that a case and controversy was created because Medicis caused Prasco to suffer an actual harm — namely, “paralyzing uncertainty” from fear that Medicis will bring an infringement suit against it.
The Federal Circuit thought differently:
To the contrary, notwithstanding this lawsuit, Prasco has launched its OSCIONâ„¢ product. More importantly, the Supreme Court has emphasized that a fear of future harm that is only subjective is not an injury or threat of injury caused by the defendant that can be the basis of an Article III case or controversy.
Rather than a purely subjective fear or the mere existence of a potentially adverse patent alone, the alleged injury at the root of most justiciable declaratory judgment controversies in the patent context is a “restraint on the free exploitation of non-infringing goods,” or an imminent threat of such restraint.
Thus, for a patentee to cause an injury they would need to, for example, create a reasonable apprehension of an infringement suit, e.g., demanding the right to royalty payments or creating a barrier to the regulatory approval of a product that is necessary for marketing.
Here, Medicis’ silence was golden.
See also:
A Hint At The Effects of MedImmune
Practical Implications of MedImmune
Supreme Court High-Fives MedImmune

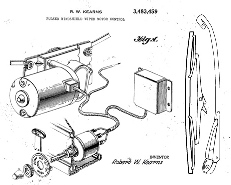 We don’t usually plug movies around here but not too many movies have patent infringement as the major plot line. Universal Pictures’ upcoming film titled Flash of Genius, starring Greg Kinnear, Lauren Graham, Dermot Mulroney and Alan Alda, tells the story of the landmark patent case of inventor Robert Kearns.
We don’t usually plug movies around here but not too many movies have patent infringement as the major plot line. Universal Pictures’ upcoming film titled Flash of Genius, starring Greg Kinnear, Lauren Graham, Dermot Mulroney and Alan Alda, tells the story of the landmark patent case of inventor Robert Kearns.
 The generic and biotech drug industries have spent a lot of cash lobbying Congress over how generic biotech drugs should be approved although McCain has
The generic and biotech drug industries have spent a lot of cash lobbying Congress over how generic biotech drugs should be approved although McCain has  Jon Dudas, Under Secretary of Commerce for Intellectual Property and Director of the United States Patent and Trademark Office has (proudly?) highlighted the fact that the allowance rate for patents is currently 42%. This is in contrast to allowance rates in excess of 70% just eight years ago. So, did patent applications really get that much worse in just a few years?
Jon Dudas, Under Secretary of Commerce for Intellectual Property and Director of the United States Patent and Trademark Office has (proudly?) highlighted the fact that the allowance rate for patents is currently 42%. This is in contrast to allowance rates in excess of 70% just eight years ago. So, did patent applications really get that much worse in just a few years?