 In an appeal from a summary judgment of noninfringement of U.S. Patent 6,054,482, the Fed Circuit concluded that the district court erred in determining that there were no genuine issues of material fact concerning whether Warner Lambert failed to meet its burden of proof that the accused products infringe the asserted claims of the ’482 patent.
In an appeal from a summary judgment of noninfringement of U.S. Patent 6,054,482, the Fed Circuit concluded that the district court erred in determining that there were no genuine issues of material fact concerning whether Warner Lambert failed to meet its burden of proof that the accused products infringe the asserted claims of the ’482 patent.
See: In Re Gabapentin Patent Litigation (involving Warner-Lambert, Pfizer and Godecke Aktiengesellschaft v. Purepac Pharma, Faulding, Watson Pharma, Danbury Pharma, Teva, Zenith Labs, Ivax, Apotex, Torpharm and Eon Labs) United States Court of Appeals for the Federal Circuit (06-1572).
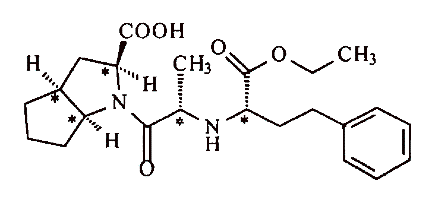
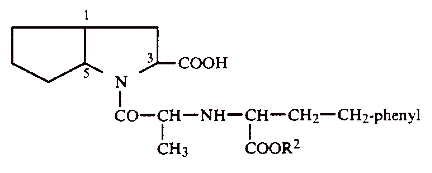
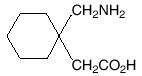
Warner Lambert sells Neurontin®, a drug used to treat certain cerebral disorders, including epilepsy. The active ingredient in Neurontin® is a compound called gabapentin, which is covered by Warner Lambert’s ’482 patent, directed towards a process for the preparation of, and compositions containing, gabapentin substantially free from a lactam contaminant.
Under certain conditions gabapentin has a tendency to form a lactam, which makes the drug unstable and unsafe. The lactam was shown to be twenty-five times more toxic than gabapentin, and is linked to causing seizures, rather than preventing them. In an effort to minimize the formation of lactam, Warner Lambert developed a process involving two limitations to achieve stable formations of gabapentin.
First, gabapentin must be highly purified before being formulated into the pharmaceutical preparation. The ’482 patent discloses that:
The active materials of formula (I) must be prepared as highly purified, nonderivatized free amino acids, for example, from the corresponding hydrochloride by ion exchange. The proportion of remaining hydrochloride admixtures should thereby not exceed 20 ppm. The same also applies to other mineral acids.
Second, certain adjuvants that reduce the stability of gabapentin must be avoided. The ’482 patent further discloses that:
The following adjuvant materials, for example, reduced the stability of the compounds (I) and should be avoided in the preparation of pharmaceutical compositions: modified maize starch, sodium croscarmelose, glycerol behenic acid ester, methacrylic acid co-polymers (types A and C), anion exchangers titanium dioxide, and silica gels such as Aerosil 200.
The district court granted summary judgment of noninfringement becuase Warner Lambert failed to adduce sufficient evidence to establish that the accused products meet the limitation that the anions of a mineral acid do not exceed 20 ppm (“the 20 ppm limitation”). Based on the undisputed fact that the test had a ± 5 ppm margin of error, the court determined that that evidence was insufficiently precise to prove infringement.
The parties gave conflicting expert opinions, based on different evidence and different methods of testing, regarding whether Teva’s samples infringed the ’482 patent. As such, Warner Lambert argues that genuine issues of material fact exist in the record, and thus summary judgment was not appropriate.
First, appellees challenged the accuracy and reliability of the pH testing method. Appellees assert that the pH testing method yielded inaccurate results because, inter alia, Warner Lambert’s expert failed to calibrate the standards used for the test. Second, appellees argued that pH testing is not competent proof of infringement in light of the test’s lack of precision.
The Federal Circuit agreed with Warner Lambert that genuine issues of material fact existed.
In moving for summary judgment of noninfringement based on Warner Lambert’s inability to meet its burden of proof, appellees informed the district court that:
It is important to note for the record that Defendants strongly dispute the capability of pH testing to make any scientifically meaningful distinctions between gabapentin samples at the trace levels of acidity relevant to the ’482 patent. However, for purposes of this motion, Defendants have placed that dispute to one side (as they must), and focused on the undisputed limitations on the precision of such comparative pH measurements.
At the summary judgment hearing, appellees expressly stated that:
Teva’s experts vehemently dispute the validity of the data [Dr. Bartlett] relied on, but I want to ignore that dispute. Those factual issues are in dispute, but should not be part of this motion, and we, you can ignore them.
Thus, appellees limited their summary judgment motion to the issue of the undisputed limits of the test’s precision, viz., the ± 5 ppm margin of error, which we have considered. As such, appellees waived any argument challenging the validity, including challenges to the accuracy or reliability, of the pH testing method for purposes of summary judgment.
Appellees also argued that the court erred in its construction of the “anion of a mineral acid” and adjuvant claim limitations, and that they should still be awarded judgment. They asserted that “anion of a mineral acid” refers to anions from any source capable of forming a mineral acid. That is, that the term refers to total chloride content and is not limited to acid-derived chloride ions.
The court stated:
We agree with the district court that the proper construction is “anion derived from a mineral acid.” “It is a ‘bedrock principle’ of patent law that ‘the claims of a patent define the invention to which the patentee is entitled the right to exclude.’” Phillips v. AWH Corp., 415 F.3d 1303, 1327 (Fed. Cir. 2005) (quoting Innova/Pure Water, Inc. v. Safari Water Filtration Sys., Inc., 381 F.3d 1111, 1115 (Fed. Cir. 2004)). Here, the plain language of the claim supports the construction that the anion specifically is derived from a mineral acid. Appellees’ assertion that the claimed anion refers to total chloride ions or anions from any source that is “capable of” forming a mineral acid is unsupported by the claim language. Had the patentees intended the anion to refer to any anion, regardless of its source, the patentees could have simply claimed “anions” and omitted the phrase “of a mineral acid.”
We have also held that claims “must be read in view of the specification, of which they are a part.” Phillips, 415 F.3d at 1315. While appellees argue that the specification provides no support for their construction, we find that the specification provides further support for the construction adopted by the district court. The specification teaches a multi-step process for making gabapentin that is substantially free from lactam.